
The U.S. healthcare system runs on generics. Right now, 9 out of 10 prescriptions filled in America are for generic drugs. That’s not luck. It’s the result of a carefully designed legal and scientific process that lets safe, effective, low-cost versions of brand-name drugs reach patients quickly. But how does the FDA actually approve these drugs? What’s the law behind it? And why does it matter to you?
The Hatch-Waxman Act: The Foundation of Generic Drug Access
The entire system hinges on one law: the Drug Price Competition and Patent Term Restoration Act of 1984. Everyone calls it the Hatch-Waxman Act, after its sponsors. Before this law, generic drug makers faced a huge barrier. To get approval, they had to run the same expensive clinical trials as the original drug company - even though the active ingredient was identical. That made generics financially impossible for most companies. Hatch-Waxman changed that. It created the Abbreviated New Drug Application, or ANDA. The word “abbreviated” is key. Generic manufacturers don’t need to prove the drug works again. They just need to prove it’s the same as the brand-name drug already approved by the FDA. This single change turned generics from a niche idea into a $125 billion industry. The law also gave brand-name companies a 5-year exclusivity period after their drug’s approval, plus up to 5 more years if they did new pediatric studies. But it balanced that with a clear path for generics to enter the market once patents expired - or if they challenged those patents legally. This balance is what keeps innovation alive while keeping prices down.What the FDA Actually Requires for Approval
The FDA doesn’t just rubber-stamp any pill that looks like the brand-name version. Every generic must meet exacting standards. Here’s what they check:- Same active ingredient: The exact chemical compound. No exceptions.
- Same strength: If the brand is 50 mg, the generic must be 50 mg - no more, no less.
- Same dosage form: Tablet, capsule, injection, patch - it has to match.
- Same route of administration: Oral, topical, inhaled - the way it enters your body must be identical.
- Same intended use: Same conditions treated, same patient population.
- Same quality standards: Made in facilities inspected to the same level as brand-name plants.
The ANDA Submission and Review Process
Applying to the FDA isn’t just filling out a form. It’s a detailed, multi-part package called an ANDA. It includes:- Chemistry, Manufacturing, and Controls (CMC) data - how the drug is made, tested, and packaged
- Facility details - every site involved must pass FDA inspection
- Proposed labeling - must match the brand’s exactly, including warnings and usage instructions
- Bioequivalence study results - with full methodology and raw data

Patents, Exclusivity, and the 30-Month Stay
Here’s where things get complicated. The Hatch-Waxman Act lets generic companies challenge brand patents. If a generic applicant files a Paragraph IV certification - meaning they believe a patent is invalid or won’t be infringed - the brand company has 45 days to sue. If they do, the FDA can’t approve the generic for up to 30 months. That’s called a 30-month stay. This is why some generics take years to hit the market, even after a patent expires. It’s not the FDA dragging its feet. It’s legal battles. Big pharma often uses multiple patents on a single drug - formulation, method of use, delivery system - to extend market protection. This tactic, called “evergreening,” is controversial but legal. The FDA has tools to fight this. They publish the Orange Book, which lists approved drugs, their patents, and exclusivity periods. Generic companies use this to plan their applications. The agency also has a Drug Competition Action Plan to remove barriers to generic entry, especially for drugs with little or no competition.Complex Generics: Where the System Gets Stuck
The ANDA pathway works beautifully for simple pills and injections. But it doesn’t work as well for complex products. Think inhalers, topical creams, long-acting injectables, or drugs that need special delivery systems. These aren’t just “copycat” versions. They require new science to prove they work the same way. For example, a generic version of an asthma inhaler isn’t just a different brand of spray. The particle size, propellant, and delivery mechanism all affect how the drug reaches the lungs. Proving bioequivalence here isn’t just about blood levels - it’s about lung deposition. The FDA calls these “complex generics,” and they’re a growing challenge. That’s why the FDA launched its Complex Generic Drug Product Development Resources initiative. They’re working with manufacturers to develop new testing methods and standards. In 2023, they approved the first generic of Vivitrol, a long-acting injectable for opioid addiction - a major milestone for complex generics.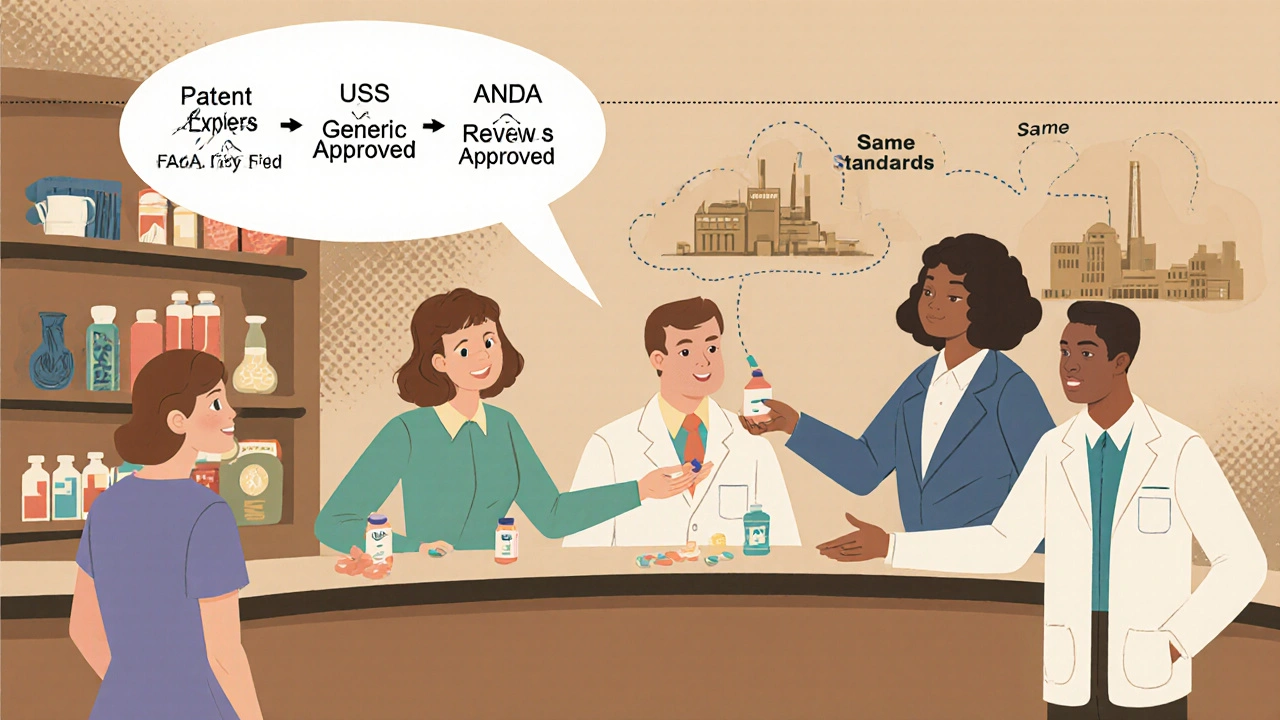
Who Makes These Drugs and Why It Matters
The generic drug market is dominated by a few big players: Teva, Sandoz, Viatris (which merged Mylan and Pfizer’s generics), and Amneal. But there are also hundreds of smaller companies that focus on niche or complex products. What’s changed in the last decade? More manufacturing is moving back to the U.S. The FDA’s October 2025 pilot program prioritizes ANDA reviews for companies that test and make their drugs domestically. That’s a direct response to supply chain risks exposed during the pandemic. If you’re buying a generic drug made in the U.S., it’s not just about cost - it’s about reliability.Why This System Works - And Why You Should Care
The FDA’s generic approval system isn’t perfect. Patent delays, complex products, and global supply chains still cause problems. But the core idea - that safe, affordable drugs should be available to everyone - has never been more important. In 2023 alone, the FDA approved 90 first-time generic drugs. That’s 90 new options for patients who might otherwise skip doses because of cost. It’s why a 30-day supply of metformin costs $4 instead of $300. It’s why people with chronic conditions can stay on treatment. This isn’t just regulatory policy. It’s public health. The FDA doesn’t just approve pills. They approve access. And that’s why the ANDA pathway remains one of the most successful public health programs in modern history.Are generic drugs as safe as brand-name drugs?
Yes. The FDA requires that generic drugs meet the same strict standards for quality, strength, purity, and performance as brand-name drugs. They’re held to the same manufacturing rules, inspected at the same facilities, and must prove bioequivalence before approval. Millions of patients use generics every day without issue.
Why do generic pills look different from brand-name pills?
Generic drugs can have different colors, shapes, or inactive ingredients like fillers or dyes because those aren’t medically active. The law only requires the active ingredient to be identical. The differences are cosmetic or for trademark reasons - they don’t affect how the drug works.
How long does it take for a generic to be approved after a patent expires?
It varies. For simple drugs with no patent challenges, approval can happen within 10 months of a clean ANDA submission. But if a brand company sues over a patent, the FDA can’t approve the generic for up to 30 months. Complex generics or those with exclusivity periods may take even longer - sometimes years.
Do all generic drugs cost 80% less than brand-name drugs?
On average, yes - generics cost 80% to 85% less. But prices can vary depending on competition. If only one generic is available, the price might be higher. Once multiple generics enter the market, prices often drop to pennies per dose. The more competition, the lower the cost.
Can I trust a generic drug made overseas?
Yes, if it’s approved by the FDA. The agency inspects manufacturing facilities worldwide - whether in the U.S., India, or China - using the same standards. The location doesn’t matter as much as whether the facility passes inspection. The FDA’s recent push for U.S.-made generics is about supply chain security, not safety.




Anne Nylander
21 November 2025 - 06:00 AM
OMG this is so cool!! I never knew generics had to pass ALL those tests!! My blood pressure med is generic and I was scared it wouldn't work but now I feel like a total genius for taking it 😍
Franck Emma
21 November 2025 - 13:21 PM
This system is a joke.
Noah Fitzsimmons
22 November 2025 - 10:16 AM
Oh wow, so the FDA just lets anyone slap a label on a pill and call it ‘bioequivalent’? And you believe that? 🙄 Tell me, when was the last time you saw a generic drug maker actually test on real patients with actual diseases? Or did they just use 24 college kids on Adderall and call it a day?
Don’t get me wrong-I love cheap meds. But this whole ‘abbreviated’ process is just corporate capitalism dressed up as public health. The brand names spent billions on R&D, and now some factory in India dumps the same powder into a blue capsule and calls it a day. And you’re okay with that?
And don’t even get me started on the ‘30-month stay.’ That’s not a legal tool-that’s a bribery loophole. Big Pharma buys time with lawsuits while patients wait to breathe. And the FDA? They’re just the bouncer at the club letting the rich kids in first.
Yeah, sure, ‘same active ingredient.’ But what about the fillers? The dyes? The coating? Ever had a generic that gave you a rash? No? Lucky you. My cousin had to switch back to brand after her generic made her dizzy for three weeks. FDA didn’t care. ‘Within 80–125%’-that’s not medicine, that’s a dice roll.
And don’t even mention ‘U.S. manufacturing.’ You think the FDA inspects every single batch? Nah. They sample. One vial out of 10,000. That’s not safety-that’s hope.
So yeah, ‘one of the most successful public health programs.’ Right. It’s successful for shareholders. Not for people who end up in the ER because their ‘equivalent’ insulin didn’t absorb right.
Call it what it is: a regulatory shell game. And we’re all the suckers paying for the tickets.
Eliza Oakes
23 November 2025 - 10:20 AM
Wait, so you’re telling me the FDA approves generics based on blood levels in healthy volunteers… but we’re supposed to trust these for people with liver disease, kidney failure, or autoimmune disorders? That’s not science-that’s assumption.
And who decided 80–125% is ‘equivalent’? That’s a 45% swing! If I took a pill that delivered 80% of the dose, I’d be underdosed. If I got 125%, I’d be overdosed. That’s not medicine, that’s a roulette wheel with your life.
Also, why do we call them ‘generics’? They’re not generic-they’re copies. And copies are never the same. Ever seen a photocopy of a photocopy? That’s your generic drug after three manufacturers have touched it.
And the ‘U.S. manufacturing push’? Cute. Most of those ‘domestic’ plants are owned by Teva or Sandoz. It’s still corporate consolidation with a red, white, and blue sticker.
And don’t even get me started on the Orange Book. It’s a maze. I’ve seen pharmacists spend hours trying to figure out which generic is actually approved for which indication. It’s not transparency-it’s obfuscation.
This whole system is held together by duct tape, hope, and FDA overtime.
Clifford Temple
24 November 2025 - 15:46 PM
Let me get this straight-we’re letting China and India make our life-saving drugs? And we’re proud of it? No wonder our veterans are getting fake insulin. This isn’t healthcare, it’s national betrayal.
The FDA’s been asleep at the wheel for 40 years. If this was cars or planes, we’d be in jail. But pills? Nah. Let’s just trust some guy in Mumbai with a lab coat and a smile.
Make generics in America or don’t make them at all. This isn’t about cost-it’s about sovereignty. And we’re losing.
Corra Hathaway
24 November 2025 - 22:29 PM
YESSSSSS!!! 🙌 This is the kind of stuff that keeps people alive and sane!! I used to skip my antidepressants because the brand was $500 a month-now I pay $4 and I’m actually functioning again 💪❤️
And the fact that the FDA checks EVERYTHING? Mind blown. I thought generics were just ‘kinda close’-but NOPE. Same active ingredient, same absorption, same everything. That’s wild!!
Also, love that they’re pushing U.S. manufacturing. More jobs + safer supply chain = WIN WIN 🇺🇸✨
PS: My grandma takes 7 generics and still hikes 5 miles every Sunday. She says, ‘If it works, why change it?’ And she’s right 😘
Nikhil Purohit
26 November 2025 - 07:29 AM
Interesting read! I’m from India, and we make a lot of these generics. But I’ve always wondered-how does the FDA ensure quality control when the manufacturing is overseas? Are inspections random or scheduled? And how often? Also, do they check the raw materials too, or just the final product?
And what happens if a batch fails inspection after approval? Do they recall it immediately or wait for complaints?
Would love to know more about the inspection protocols. Thanks for sharing this!
Debanjan Banerjee
27 November 2025 - 22:23 PM
Actually, the FDA doesn’t just inspect manufacturing sites-they do unannounced audits, and they’ve been ramping up inspections in India and China since 2018. In 2022, over 50% of all ANDA inspections were overseas, and 92% passed. That’s higher than the domestic pass rate.
Raw materials are tracked via supply chain documentation and verified through third-party lab testing. If a batch fails, the FDA can issue an Import Alert, which blocks all future shipments from that facility until they fix it. No warning. No grace period.
And yes-they check every active pharmaceutical ingredient (API) source, not just the final pill. That’s why some generics get pulled months after approval. It’s not incompetence-it’s diligence.
Also, the 80–125% bioequivalence range? That’s based on pharmacokinetic variability in healthy populations. For drugs with narrow therapeutic windows (like warfarin or levothyroxine), the FDA requires tighter ranges-sometimes 90–111%.
So yes, you can trust them. But you’re right to ask. That’s how systems improve.